In protein engineering, site-directed mutagenesis methods are used to generate DNA sequences with mutated codons, insertions or deletions. In a widely used method, mutations are generated by PCR using a pair of oligonucleotide primers designed with mismatching nucleotides at the center of the primers. In this method, primer-primer annealing may prevent cloning of mutant cDNAs. To circumvent this problem we developed an alternative procedure that does not use forward-reverse primer pair in the same reaction.
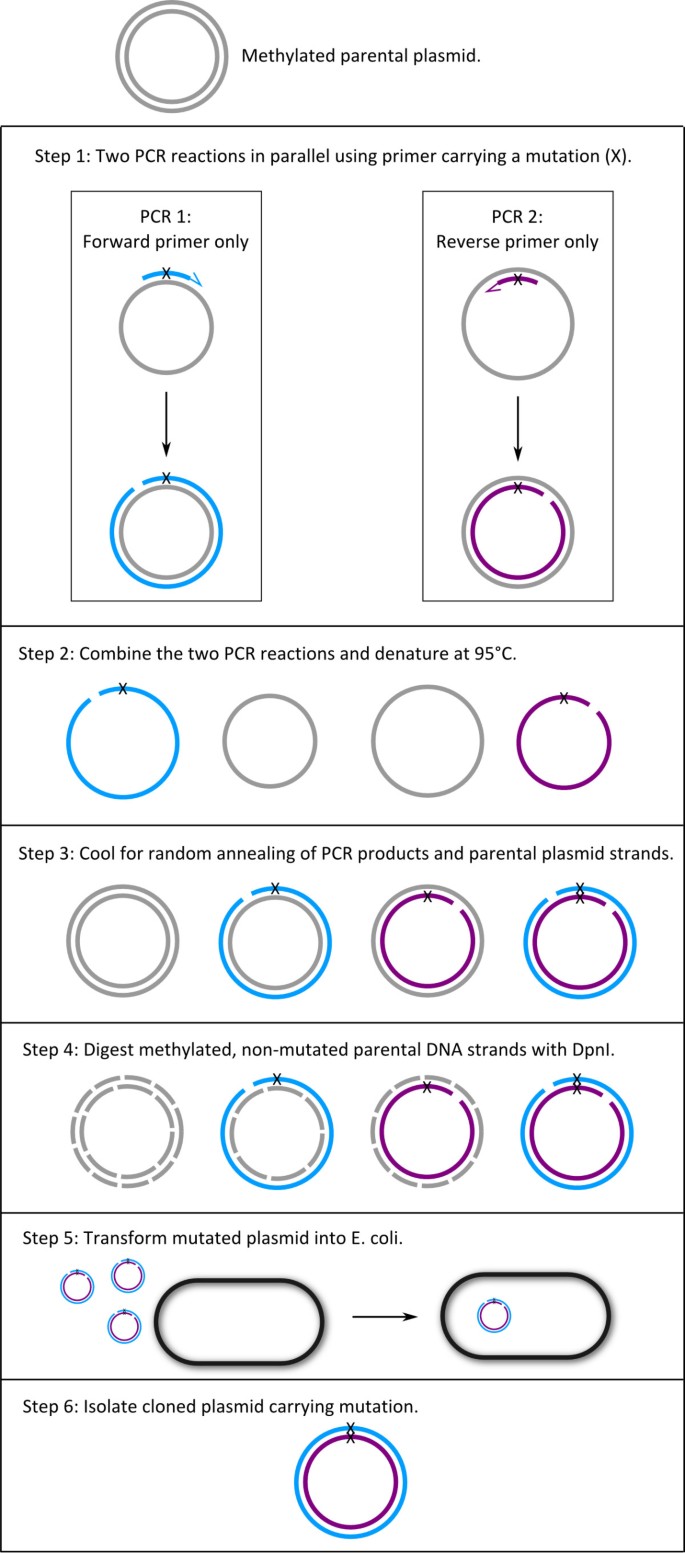
In initial studies we used a double-primer PCR mutagenesis protocol, but sequencing of products showed tandem repeats of primer in cloned DNA. We developed an alternative method that starts with two Single-Primer Reactions IN Parallel using high-fidelity Pwo DNA polymerase. Thus, we call the method with the acronym SPRINP. The SPRINP reactions are then combined, denatured at 95°C, and slowly cooled, promoting random annealing of the parental DNA and the newly synthesized strands. The products are digested with DpnI that digests methylated parental strands, and then transformed into E. coli. Using this method we generated >40 mutants in cDNAs coding for human Epithelial Na + Channel (ENaC) subunits. The method has been tested for 1–3 bp codon mutation and insertion of a 27 bp epitope tag into cDNAs.
The SPRINP mutagenesis protocol yields mutants reliably and with high fidelity. The use of a single primer in each amplification reaction increases the probability of success of primers relative to previous methods employing a forward and reverse primer pair in the same reaction.
Site-directed mutagenesis (SDM) methods are used to generate cloned DNAs with modified sequences for examining the importance of specific residues in protein structure and function. SDM represents the primary rational method in protein engineering and for altering enzyme substrate selectivity [1, 2].
Currently prevalent methods of SDM employ PCR using oligonucleotide primer pairs that carry the desired mutation. There is a large variety of approaches for SDM using PCR [1, 3, 4]. One of the widely used methods is QuikChange [5]. This method and its later modifications employ complementary primer pairs in the same PCR reaction. The use of complementary primer pairs may lead to the formation of "primer dimers" by partial annealing of a primer with the second primer in reaction, and formation of tandem repeats of primers, reducing the yield of successful transformants. Primer-primer annealing is especially severe in SDM because the primers used include mismatching nucleotides to generate the desired mutation. To avoid these problems various alternative protocols have been developed [6, 7]. Similarly, in our research on the structure-function relationship of human epithelial sodium channel (ENaC) subunits we failed to generate desired clones using a commercial site-directed mutagenesis kit employing double-primers reactions.
We developed a SDM method that includes two PCR reactions run in parallel with each one of the forward and reverse primers. At the end of the PCR, the reactions are combined and after three steps of denaturation, renaturation and DpnI digestion of methylated parental plasmid DNA, the products are directly transformed into host cells.
We cloned the cDNAs encoding for the three subunits of human Epithelial Na + Channel (α, β and γ ENaC) [8–10] for analyzing structure-function relationships in these proteins. The α, β and γ ENaC cDNA inserts (2013, 1923 and 1950 bp, respectively) were cloned in the expression plasmid pGEM-HJ. The plasmid pGEM-HJ is about 3,000 bp long and includes a T7 promoter used for transcription of cRNA for micro-injection and expression of proteins in Xenopus oocytes. Since the method requires methylated parental plasmid DNA, all plasmids used in these studies were isolated from XL1-Blue cells (Stratagene) that have a dam+ (wild type) genotype encoding Dam methylase.
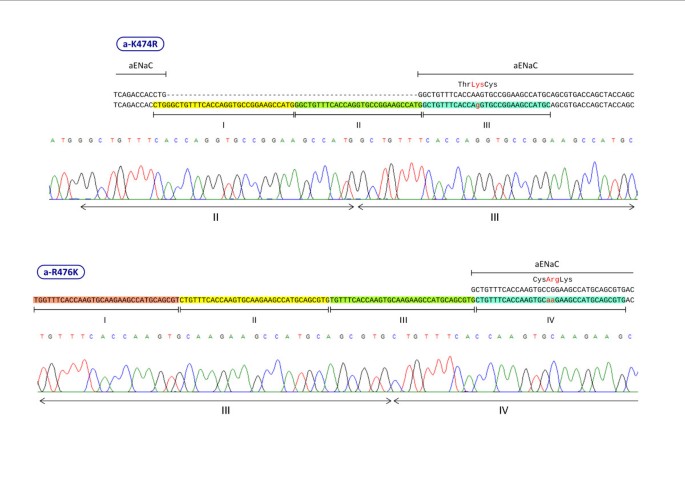
We designed primers for two types of mutagenesis: 1. Mutation of 1–3 nucleotides to change a codon sequence; 2. Insertion of a 27 bp segment for adding a hemagglutinin (HA) peptide epitope tag into cDNA inserts. Table 1 includes seven sets of representative primer sequences we used for site-directed mutagenesis. Forward and reverse primers were designed using guidelines similar to that commonly employed in double-primer PCR reactions:
For SDM of a cDNA, we carry out two PCR reactions in parallel with the forward and the reverse primers in separate tubes (see Table 2 for reaction components and concentrations). After the initial denaturation step at 94°C for 2 min, PCR was conducted for 30 cycles with denaturation at 94°C for 40 s, primer annealing at 55°C for 40 s and DNA synthesis at 72°C for 60 s for each 1 kb of cDNA + plasmid sequence, e. g. 5 min for a plasmid + cDNA sequence of 5 kb. The DNA polymerase we use is Pwo (Roche, Germany).
To overcome the problem of primer-primer annealing observed with the double primer-procedure, we developed an alternative single-primer PCR procedure outlined in Figure 1. We first tested this protocol using the same primer-sets that did not work with the double-primer method. Seeing the success of the protocol we continued to employ it to generate all the mutants needed for our studies.
We start the single-primer PCR protocol with ~500 ng plasmid template that is about 10 times higher than that recommended for the double-primer PCR (Table 4) because, DNA amplification in the single-primer PCR is much lower (Table 4). After each transformation we obtained consistently several hundred colonies. We routinely checked five colonies at random from each transformation by direct sequencing of the plasmid. In 26 transformations that we carried out, the number of plasmids with the designed mutated sequence averaged 3.6 (range: 2–5) out of 5 plasmids from 5 independent colonies. Plasmids that did not have the expected mutated sequence had the original sequence without a mutation. So far, we have not observed tandem repeats of a primer as we observed in the double-primer PCR method (Figure 2).
In 26 SDM experiments, the length of the primers ranged between 31–36 nt. The range for Tm of the primers was 79–92°C and the range for percentage of G+C was 44–76%. These ranges slightly exceed the primer-design guidelines noted in Methods. Since each mutation has to be located at a specific position, we could not always find a suitable primer sequence within the guidelines. Although, we exceeded slightly the guideline range, all the primers worked successfully.
If we define "success" as finding a correctly mutated cDNA among 5 sample colonies examined, then our success rate has been 100% for all single-primer mutation experiments we have carried out. The method has been used in two other laboratories (Prof. Nathan Dascal and Prof. Ilana Lotan, Tel-Aviv University) with a similar success rate of 100%.
The agarose gel in Figure 3 shows the DNA products of one sample at successive steps of our procedure. Plasmid alone, prior to PCR, shows two major bands. After PCR with forward (F) or reverse (R) primers, two major bands can be seen that correspond to parental plasmid DNA. In addition, a new band at the expected size of ~5 kb appears, representing the PCR synthesized linear DNA that includes the cDNA insert and the plasmid. Additional smaller bands represent non-specific PCR products.
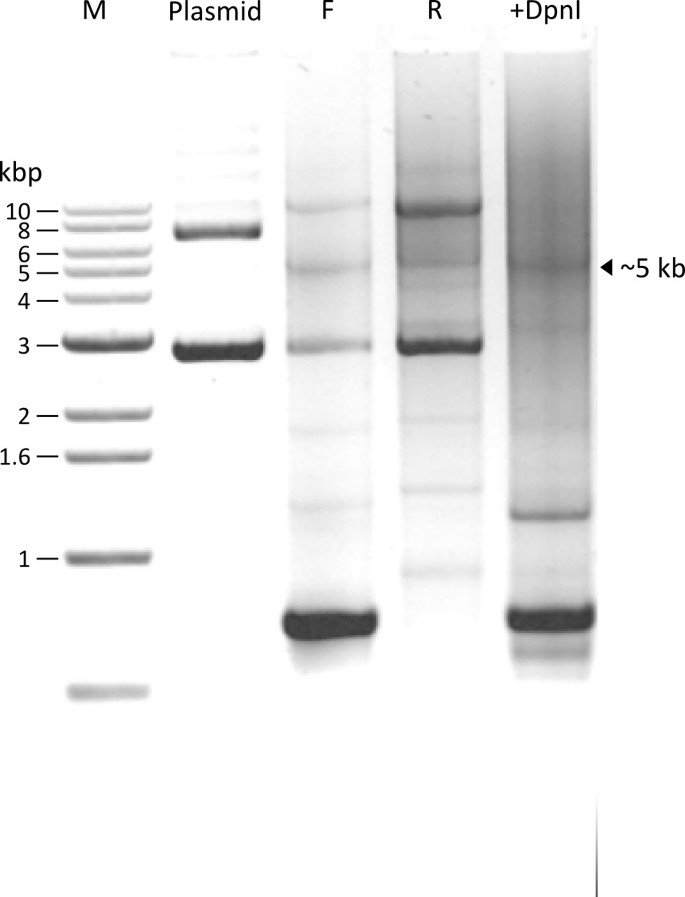
Since each PCR reaction is carried out with only one primer, amplification of the DNA is not high (only 30 fold) as compared to the double-primer PCR procedure (Table 4). Therefore, the newly synthesized full size linear plasmid strands do not appear as strong bands on the agarose gel. After DpnI digestion, the two major bands present in the Plasmid lane and in the F and R primer lanes disappear (+DpnI lane), confirming that these two bands represent methylated parental DNAs that were digested by DpnI.
In routine application of our parallel PCR SDM method, we carry out all steps of the method successively without visualizing intermediate products during the whole procedure, i.e., we do not run a gel to examine PCR products. We combine the contents of the two PCR tubes, and proceed with the denaturation and reannealing protocol in Table 3. After this step the DNA is directly taken for transformation into competent E. coli cells.
Site-directed mutagenesis methods are crucial in analyzing structure-function relationships. But, the large number of methods published in the literature attest to the difficulty of executing these methods reliably and efficiently. We observed that widely used double-primer protocol resulted in insertion of multiple copies of primer probably due to primer-primer annealing (Figure 2). The site-directed mutagenesis method we present here circumvents the use of primer pairs in the same PCR. Thus, we call it with the acronym SPRINP (Single-Primer Reactions IN Parallel). We have tested the method in our studies on ENaC subunits, and we have also submitted the method for independent testing by two other laboratories as noted in the Results. So far we have observed no instance of multiple primers in the clones we have isolated.
Table 4 provides a list of the differences between the single-primer PCR and the double-primer PCR methods. Probably a most important factor in the success of our method is that we carry out the initial mutagenesis with a single primer in each reaction. In SDM protocols that use forward and reverse primers with complementary sequences, primer-primer annealing can be a significant problem as we observed in our use of double-primer method.
In insertion mutagenesis longer primers with long mismatching segments are used, thus enhancing the probability of non-specific reactions [1]. As noted in Table 1, we used successfully a 57 nt primer (with a 27 nt insertion that did not match the parental strand) in an experiment to introduce an epitope tag into β ENaC cDNA.
In the current study we have introduced mutations and insertions at only one point. Some structure-function studies require multiple-site mutagenesis [12]. We have not examined the use of multiple primers to generate multiple-site mutations. However, the basic principle of the method can be applied for use with multiple primers for multiple-site mutagenesis.
Using the Single-Primer Reactions IN Parallel (SPRINP) mutagenesis protocol that we developed we could generate cDNAs with point mutations and insertions reliably and with high fidelity. The use of a single primer in each amplification reaction increases the probability of successful use of primers relative to previous methods employing a forward and reverse primer pair in the same reaction.
This research was funded in part by a grant from the Chief Scientist Office of the Israel Ministry of Health.